a:2:{s:4:"TEXT";s:65762:"
Галлервордена-Шпатца (Пантотенат-киназа-ассоциированная нейродегенерация, PKAN): Комплексный обзор
Нейродегенерация, связанная с пантотенат-киназой (Пантотенат-киназа-ассоциированная нейродегенерация, PKAN), ранее известная как болезнь Галлервордена-Шпатца, является редким, прогрессирующим, генетически обусловленным заболеванием, характеризующимся накоплением железа в базальных ганглиях головного мозга. Это состояние относится к группе наследственных нарушений обмена веществ, известных как нейродегенерация с накоплением железа в головном мозге (NBIA). Переименование заболевания было обусловлено этическими соображениями, связанными с ролью докторов Юлиуса Галлервордена и Хуно Шпатца в нацистской Германии, и фокусировкой на генетической основе заболевания, что подчеркивает важность точной номенклатуры в медицине [1]. Данный обзор призван предоставить актуальную и всестороннюю информацию о PKAN, охватывая этиологию, патогенез, клиническую картину, диагностику, лечение и прогноз, опираясь на современные научные данные.
Определение и МКБ-10
Пантотенат-киназа-ассоциированная нейродегенерация (PKAN) — это редкое аутосомно-рецессивное нейродегенеративное заболевание, вызванное мутациями в гене *PANK2*. Оно характеризуется прогрессирующей дистонией, паркинсонизмом, пирамидными симптомами, дизартрией, дисфагией, а также когнитивными и психиатрическими нарушениями. Главной патологической особенностью PKAN является избыточное накопление железа в базальных ганглиях, особенно в бледном шаре, что визуализируется при магнитно-резонансной томографии (МРТ) головного мозга [2].
PKAN представляет собой генетически детерминированное заболевание с характерным паттерном отложения железа в центральной нервной системе, что обуславливает специфическую клиническую картину.
В Международной классификации болезней 10-го пересмотра (МКБ-10) PKAN классифицируется под кодом **G23.0** – Нейродегенерация, связанная с накоплением железа в головном мозге (Neurodegeneration with brain iron accumulation, NBIA), или ранее использовался код **G23.0** (Болезнь Галлервордена-Шпатца), который теперь чаще заменяется на более точное описание в контексте NBIA.
Точная классификация PKAN в МКБ-10 позволяет стандартизировать медицинскую статистику и облегчает взаимодействие между специалистами в области редких заболеваний.
Этиология
Основной причиной PKAN являются биаллельные мутации в гене *PANK2*, расположенном на хромосоме 20p13 [3]. Ген *PANK2* кодирует белок пантотенат-киназу 2, один из четырех изоферментов пантотенат-киназы. Пантотенат-киназа играет ключевую роль в первом и скорость-лимитирующем этапе биосинтеза кофермента А (КоА) из пантотената (витамина B5). Это фермент, который катализирует фосфорилирование пантотената до 4'-фосфопантотената.
Мутации в гене *PANK2* приводят к дефициту функциональной пантотенат-киназы 2, нарушая критический путь синтеза кофермента А.
PKAN наследуется по аутосомно-рецессивному типу. Это означает, что для развития заболевания человек должен унаследовать две копии мутированного гена *PANK2* – по одной от каждого родителя, которые являются бессимптомными носителями [4]. Если человек наследует только одну мутированную копию, он является носителем, но не проявляет симптомов заболевания.
Аутосомно-рецессивный тип наследования обуславливает появление заболевания только у индивидов, получивших патологические аллели от обоих родителей, что имеет важное значение для генетического консультирования.
Эпидемиология
PKAN является одним из наиболее распространенных типов нейродегенерации с накоплением железа в головном мозге (NBIA), составляя примерно 30-50% всех случаев NBIA [5]. Однако, даже с учетом этого, PKAN остается чрезвычайно редким заболеванием. Его распространенность оценивается примерно в 1-3 случая на 1 000 000 человек в общей популяции.
Несмотря на то, что PKAN является самым частым подтипом NBIA, его общая распространенность крайне низка, подтверждая статус орфанного заболевания.
Заболевание встречается по всему миру, без выраженной этнической или географической предрасположенности, хотя в некоторых популяциях могут наблюдаться так называемые "эффекты основателя" (founder effect), приводящие к повышенной частоте встречаемости определенных мутаций [6]. Мужчины и женщины страдают одинаково часто. Возраст дебюта и тяжесть симптомов значительно варьируются, что объясняет наличие классической и атипичной форм заболевания.
Глобальное распространение PKAN и отсутствие выраженной гендерной предрасположенности подчеркивают случайный характер возникновения мутаций и важность универсальных скрининговых подходов для редких генетических заболеваний.
.png")
Патогенез
Патогенез PKAN сложен и не до конца изучен, но центральную роль играет дефицит функционального белка пантотенат-киназы 2. Этот фермент необходим для первого шага синтеза КоА, который является критическим кофактором для сотен ферментов, участвующих в метаболизме жирных кислот, цикла Кребса, синтезе гема, детоксикации и многих других важнейших клеточных процессах [7]. Нарушение синтеза КоА приводит к накоплению промежуточных метаболитов и нарушению нормального метаболизма.
Нарушение функции пантотенат-киназы 2 критически сказывается на биосинтезе КоА, что приводит к широкому спектру метаболических дисфункций в клетках, особенно в нейронах.
**Важная информация о патогенезе:** Ключевым звеном патогенеза является нарушение гомеостаза железа. Точный механизм его накопления остается предметом исследований, но предполагается, что дефицит КоА может нарушать функцию митохондрий, выработку АТФ и регуляцию окислительного стресса, что в конечном итоге приводит к изменению метаболизма железа в нейронах. Избыток свободного железа в базальных ганглиях способствует образованию свободных радикалов, вызывая окислительный стресс и повреждение липидов, белков и ДНК, что приводит к гибели нейронов и прогрессирующей нейродегенерации [8]. Это особенно заметно в бледном шаре (globus pallidus) и черной субстанции.
Накопление железа в базальных ганглиях, вызванное нарушением КоА-зависимых процессов, и последующий окислительный стресс являются основными факторами нейродегенерации при PKAN.

В результате дефицита КоА может происходить накопление цистеина и производных цистеина, которые могут образовывать комплексы с железом, способствуя его осаждению в тканях. Это также может привести к изменению липидного состава клеточных мембран и нарушению миелинизации, усугубляя нейрональное повреждение.
Помимо прямого влияния на метаболизм железа, дефицит КоА при PKAN может косвенно способствовать накоплению цистеина и нарушению миелинизации, усиливая нейродегенеративный процесс.
Классификация
PKAN традиционно подразделяется на две основные формы в зависимости от возраста дебюта и темпов прогрессирования: классическую (типичную) и атипичную [9].
Классическая форма (Classic PKAN)
Эта форма составляет около 75-80% всех случаев PKAN. Дебют симптомов обычно приходится на раннее детство, чаще всего до 6 лет. Заболевание характеризуется быстрым прогрессированием и более тяжелым течением.
Классическая форма PKAN является наиболее распространенной и проявляется в раннем детстве с быстрым нарастанием неврологических симптомов.
Атипичная форма (Atypical PKAN)
Атипичная форма составляет оставшиеся 20-25% случаев. Симптомы обычно проявляются в позднем детстве, подростковом возрасте или даже во взрослом возрасте (после 10 лет). Прогрессирование заболевания при атипичной форме значительно медленнее, чем при классической, и клиническая картина может быть менее выраженной или включать более значимые психиатрические компоненты.
Атипичная форма PKAN характеризуется более поздним началом и медленным прогрессированием, часто с выраженными психиатрическими проявлениями, что усложняет раннюю диагностику.
Клиническая картина
Клиническая картина PKAN характеризуется прогрессирующими двигательными нарушениями, которые могут сопровождаться когнитивными и психиатрическими расстройствами.
Классическая форма PKAN
Симптомы проявляются в раннем детстве, обычно между 3 и 6 годами. Начало часто внезапное.
- Прогрессирующая дистония: Часто является первым и наиболее выраженным симптомом, затрагивая конечности, туловище, шею (тортиколис) и лицевую мускулатуру (блефароспазм, оромандибулярная дистония).
- Паркинсонизм: Ригидность, брадикинезия (замедленность движений), тремор покоя.
- Пирамидные симптомы: Повышение мышечного тонуса (спастичность), гиперрефлексия, патологические рефлексы.
- Дисфагия: Затруднение глотания, что может приводить к аспирации и недоеданию.
- Дизартрия: Нарушение артикуляции речи, часто из-за дистонии оромандибулярной области.
- Хореоатетоз: Непроизвольные, нерегулярные, быстрые движения конечностей и туловища.
- Ретинальная дегенерация: Может проявляться пигментным ретинитом, ведущим к прогрессирующему снижению зрения.
- Когнитивные нарушения: Могут быть от легких до умеренных, включая задержку развития и снижение интеллектуальных функций.
Классическая PKAN характеризуется ранним началом и быстрым прогрессированием тяжелых двигательных расстройств, таких как дистония и паркинсонизм, с возможным вовлечением зрения и когнитивных функций, что значительно ухудшает качество жизни пациентов [10].
Атипичная форма PKAN
Симптомы обычно проявляются в более позднем возрасте, после 10 лет, часто в подростковом или молодом взрослом возрасте. Прогрессирование заболевания медленнее, а клиническая картина может быть более вариабельной.
Атипичная PKAN характеризуется более поздним и медленным прогрессированием, часто с акцентом на психиатрические симптомы и дизартрию, а двигательные расстройства могут быть менее генерализованными [11].
**Ключевые клинические проявления PKAN включают прогрессирующую дистонию, паркинсонизм и когнитивные нарушения, тяжесть и возраст дебюта которых определяют форму заболевания.**
Методы диагностики
Диагностика PKAN основывается на комбинации клинической картины, характерных изменений при нейровизуализации и подтверждении диагноза с помощью генетического тестирования.
Клиническая оценка и анамнез
Подробный сбор анамнеза о развитии симптомов, возрасте их начала, темпах прогрессирования и наличии подобных случаев в семье крайне важен. Неврологический осмотр выявляет характерные двигательные расстройства (дистонию, паркинсонизм, спастичность), нарушения речи и глотания, а также когнитивные дефициты.
Тщательная клиническая оценка и сбор анамнеза являются первым шагом в диагностике PKAN, позволяя заподозрить заболевание на основе характерных неврологических проявлений.
Нейровизуализация (МРТ головного мозга)
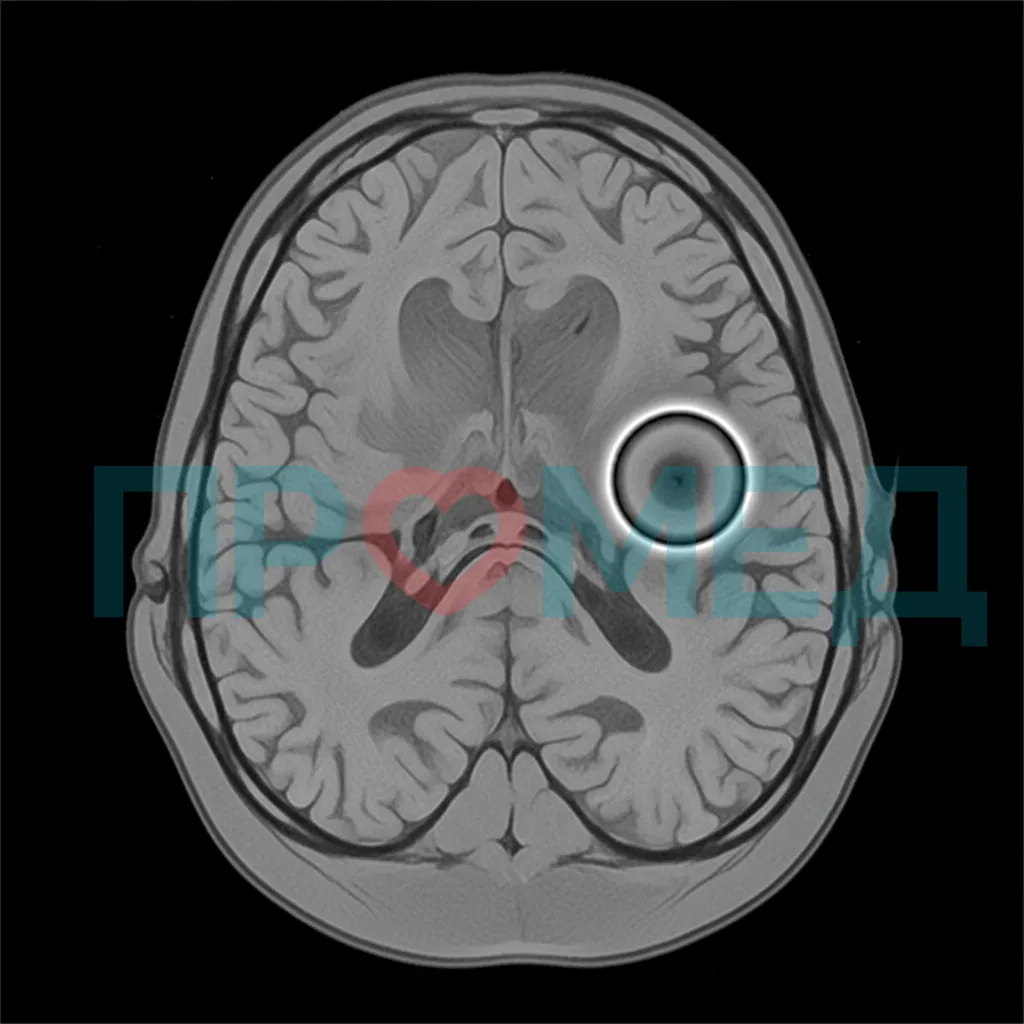
Магнитно-резонансная томография (МРТ) головного мозга является критически важным методом диагностики PKAN. Характерным признаком является так называемый **"глаз тигра" (eye-of-the-tiger sign)**, который наблюдается на Т2-взвешенных изображениях в бледном шаре. Этот признак представляет собой центральную зону повышенной интенсивности сигнала (из-за глиоза и нейрональной потери) в окружении зоны низкой интенсивности сигнала (из-за накопления железа) [12]. Он присутствует примерно у 90% пациентов с классической формой PKAN и значительно реже при атипичной форме.
МРТ головного мозга с характерным "глазом тигра" является высокоспецифичным признаком PKAN, указывающим на накопление железа в бледном шаре и помогающим отличить его от других нейродегенеративных заболеваний.
Генетическое тестирование
Подтверждающим методом диагностики является генетическое тестирование на мутации в гене *PANK2*. Секвенирование следующего поколения (NGS) или секвенирование по Сэнгеру позволяют выявить биаллельные патогенные мутации в *PANK2* [13].
Генетическое тестирование на мутации в гене *PANK2* является золотым стандартом для окончательного подтверждения диагноза PKAN.
Лабораторные исследования
Рутинные лабораторные анализы крови и мочи обычно не выявляют специфических изменений, характерных для PKAN. Однако они могут быть полезны для исключения других метаболических или нейродегенеративных заболеваний, таких как болезнь Вильсона (путем измерения уровня церулоплазмина и меди) [14].
Лабораторные исследования служат для исключения других заболеваний со схожей клинической картиной, подтверждая уникальность PKAN.
Электрофизиологические исследования
Электромиография (ЭМГ) и исследование нервной проводимости (ЭНМГ) обычно нормальные, если нет сопутствующих невропатий. Зрительные вызванные потенциалы (ЗВП) могут показать отклонения при наличии ретинальной дегенерации или атрофии зрительного нерва.
Электрофизиологические исследования могут быть полезны для оценки специфических неврологических нарушений, таких как зрение, но не являются диагностическими для PKAN сами по себе.
Дифференциальный диагноз
Дифференциальная диагностика PKAN включает широкий спектр заболеваний, характеризующихся двигательными расстройствами, когнитивными нарушениями и/или накоплением железа в головном мозге.
- **Другие формы NBIA (Нейродегенерация с накоплением железа в головном мозге)**:
- **PLA2G6-ассоциированная нейродегенерация (PLAN)**: ранняя и поздняя формы, с гипомиелинизацией и атрофией мозжечка.
- **BPAN (Beta-propeller protein-associated neurodegeneration)**: с Х-сцепленным наследованием, характеризуется задержкой развития, двигательными нарушениями и атипичным паркинсонизмом в зрелом возрасте.
- **Cystosilicosis with neurodegeneration (CN)**: характеризуется накоплением цистина, а не железа.
- **FAHN (Fatty acid hydroxylase-associated neurodegeneration)**: с аутосомно-рецессивным наследованием и спастической параплегией.
- **Болезнь Вильсона**: Наследственное нарушение обмена меди, также проявляющееся двигательными расстройствами и психическими нарушениями. Диагностируется по снижению церулоплазмина, повышению меди в моче и специфическим изменениям на МРТ (отсутствие "глаза тигра").
- **Ювенильная форма болезни Гентингтона**: Характеризуется хореей, дистонией, когнитивными и психиатрическими расстройствами, но имеет аутосомно-доминантный тип наследования и выявляется по экспансии CAG-повторов в гене *HTT*.
- **Нейроаксониальная дистрофия младенческого типа (INAD)**: Раннее начало, дистония, гипотония, задержка развития. Отсутствие накопления железа.
- **Атипичная дистония-паркинсонизм с ранним началом**: Например, связанная с мутациями в гене *GCH1* (DOPA-респонсивная дистония).
- **Миоклонус-дистония**: Может имитировать некоторые двигательные расстройства.
Комплексный дифференциальный диагноз PKAN требует тщательного анализа клинических данных, результатов нейровизуализации и генетического тестирования для исключения других нейродегенеративных и метаболических заболеваний с похожими проявлениями [15].
Сравнительная таблица дифференциальной диагностики
|
Признак/Заболевание
|
PKAN
|
Болезнь Вильсона
|
Другие формы NBIA (напр., PLAN, BPAN)
|
|
**Причина**
|
Мутации в *PANK2*
|
Мутации в *ATP7B*
|
Мутации в *PLA2G6*, *WDR45* и др.
|
|
**Патология**
|
Накопление железа в бледном шаре
|
Накопление меди в базальных ганглиях, печени, роговице
|
Накопление железа в разных отделах мозга, иногда с гипомиелинизацией
|
|
**Тип наследования**
|
Аутосомно-рецессивный
|
Аутосомно-рецессивный
|
Различный (аутосомно-рецессивный, Х-сцепленный)
|
|
**МРТ головного мозга**
|
"Глаз тигра" в бледном шаре (Т2-взвешенные изображения)
|
Гиперинтенсивные сигналы в базальных ганиях, таламусе, стволе мозга (Т2); отсутствие "глаза тигра"
|
Различные паттерны накопления железа, гипомиелинизация
|
|
**Лабораторные показатели**
|
Неспецифичны
|
Снижение церулоплазмина, повышение меди в моче, кольца Кайзера-Флейшера
|
Неспецифичны (иногда)
|
|
**Клинические проявления**
|
Дистония, паркинсонизм, дизартрия, дисфагия, психиатрические расстройства, ретинальная дегенерация
|
Двигательные расстройства, психиатрические нарушения, печеночная недостаточность, дистония
|
Различные неврологические и системные проявления
|
|
**Возраст дебюта**
|
Детство/подростковый/взрослый
|
Детство/подростковый/взрослый
|
Различный (от младенчества до взрослого)
|
Методы лечения
В настоящее время не существует этиологического или патогенетического лечения, способного остановить прогрессирование PKAN. Терапия направлена на облегчение симптомов и улучшение качества жизни пациентов.
Симптоматическая терапия
Симптоматическое лечение является основой управления PKAN и включает междисциплинарный подход.
- **Для дистонии**:
- **Инъекции ботулинического токсина**: Эффективны для уменьшения локализованной дистонии (например, тортиколиса, блефароспазма, оромандибулярной дистонии) [16].
- **Пероральные препараты**: Антихолинергические средства (например, тригексифенидил), баклофен (перорально или интратекально), бензодиазепины (например, клоназепам) могут помочь уменьшить мышечный тонус и спазмы.
- **Для паркинсонизма**:
- **Леводопа**: Может быть эффективна у некоторых пациентов, особенно на ранних стадиях, но эффект часто бывает временным [17].
- **Для других симптомов**:
- **Физиотерапия, эрготерапия, логопедия**: Важны для поддержания подвижности, улучшения равновесия, адаптации повседневной деятельности, а также для коррекции нарушений речи и глотания [18].
- **Гастростомия**: Может потребоваться при тяжелой дисфагии для обеспечения адекватного питания и предотвращения аспирации.
- **Психиатрическая поддержка и медикаменты**: Для лечения депрессии, тревоги, ОКР и других психических расстройств.
Симптоматическая терапия PKAN направлена на максимальное облегчение двигательных и других расстройств с использованием широкого спектра фармакологических и нефармакологических методов, улучшая качество жизни пациентов.
Исследуемые методы терапии
Ведутся активные исследования новых подходов к лечению PKAN, направленных на патогенетические механизмы.
- **Хелаторы железа (Deferiprone)**: Деферипрон является пероральным хелатором железа, который способен проникать через гематоэнцефалический барьер и связывать избыточное железо в мозге. Предварительные исследования показали некоторое замедление прогрессирования заболевания и даже улучшение некоторых двигательных симптомов у части пациентов [19]. Однако результаты крупных рандомизированных контролируемых исследований неоднозначны, и его применение пока не одобрено повсеместно для лечения PKAN.
Деферипрон является перспективным, но пока экспериментальным методом лечения PKAN, направленным на снижение уровня железа в мозге, но его клиническая эффективность требует дальнейших подтверждений.
- **Пантотенат (витамин B5) и его производные**: Некоторые исследования изучают возможность использования высоких доз пантотената или его аналогов для обхода дефицита пантотенат-киназы и восстановления синтеза КоА [20]. Пока нет убедительных данных о клинической эффективности такого подхода.
- **Генная терапия**: Находится на доклинической стадии разработки. Цель – введение функциональной копии гена *PANK2* в клетки мозга для восстановления нормального уровня пантотенат-киназы.
- **DBS (Глубокая стимуляция мозга)**: В некоторых случаях тяжелой дистонии у пациентов с PKAN DBS показала потенциал для улучшения двигательных симптомов, однако это инвазивная процедура с индивидуально оцениваемыми рисками и преимуществами [21].
Разработка новых терапевтических стратегий для PKAN, включая хелаторы железа, заместительную терапию пантотенатом и генную терапию, активно ведется, но большинство из них пока находятся на стадии исследований.
**Важно: В настоящее время не существует этиологического лечения PKAN, терапия направлена на облегчение симптомов и поддержание качества жизни, а большинство патогенетических методов находятся на стадии исследований.**
Клинические рекомендации
Ведение пациентов с PKAN требует мультидисциплинарного подхода с участием неврологов, генетиков, реабилитологов, диетологов, офтальмологов, психиатров и социальных работников.
- **Регулярный мониторинг**: Необходим для оценки прогрессирования заболевания, эффективности лечения и выявления новых симптомов.
- **Индивидуальный план реабилитации**: Должен разрабатываться для каждого пациента с учетом его специфических потребностей.
- **Психологическая и социальная поддержка**: Крайне важна как для пациентов, так и для их семей, сталкивающихся с хроническим прогрессирующим заболеванием.
- **Генетическое консультирование**: Должно быть предложено семьям для оценки рисков повторения заболевания и планирования семьи.
Мультидисциплинарный подход, регулярный мониторинг и всесторонняя поддержка являются краеугольными камнями комплексного управления PKAN, направленного на оптимизацию ухода за пациентами.

Реабилитация
Реабилитация при PKAN играет жизненно важную роль в поддержании функций, минимизации осложнений и улучшении качества жизни, несмотря на прогрессирующий характер заболевания.
Физическая реабилитация
Включает лечебную физкультуру (ЛФК), пассивные и активные упражнения для поддержания мышечной силы, предотвращения контрактур и улучшения гибкости. Физиотерапевты работают над улучшением походки, равновесия и координации, что особенно важно при прогрессировании дистонии и паркинсонизма [22].
Физическая реабилитация помогает замедлить потерю двигательных функций, предотвратить осложнения и поддерживать максимально возможную мобильность у пациентов с PKAN.
Эрготерапия
Эрготерапевты помогают пациентам адаптировать повседневную деятельность (одевание, еда, личная гигиена) с использованием вспомогательных средств и модификаций окружающей среды. Это способствует сохранению независимости и автономии пациента на как можно более длительный срок.
Эрготерапия фокусируется на адаптации среды и навыков для поддержания самостоятельности пациентов в выполнении повседневных задач, улучшая их функциональный статус.
Логопедия
Логопедическая помощь необходима для пациентов с дизартрией и дисфагией. Занятия включают упражнения для улучшения артикуляции, голоса, а также техники для безопасного глотания, что помогает предотвратить аспирационную пневмонию и улучшить нутритивный статус [23].
Логопедическая коррекция является важным компонентом реабилитации, направленным на улучшение коммуникативных функций и обеспечение безопасного питания у пациентов с PKAN.
Психологическая поддержка
Психологическая помощь необходима как пациентам, так и их семьям для coping с хроническим прогрессирующим заболеванием. Психотерапия, консультирование и группы поддержки помогают справиться с депрессией, тревогой, чувством потери и поддерживают качество жизни.
Психологическая поддержка является неотъемлемой частью комплексной реабилитации, обеспечивая эмоциональное благополучие и адаптацию к жизни с хроническим заболеванием для пациентов и их семей.
Прогноз
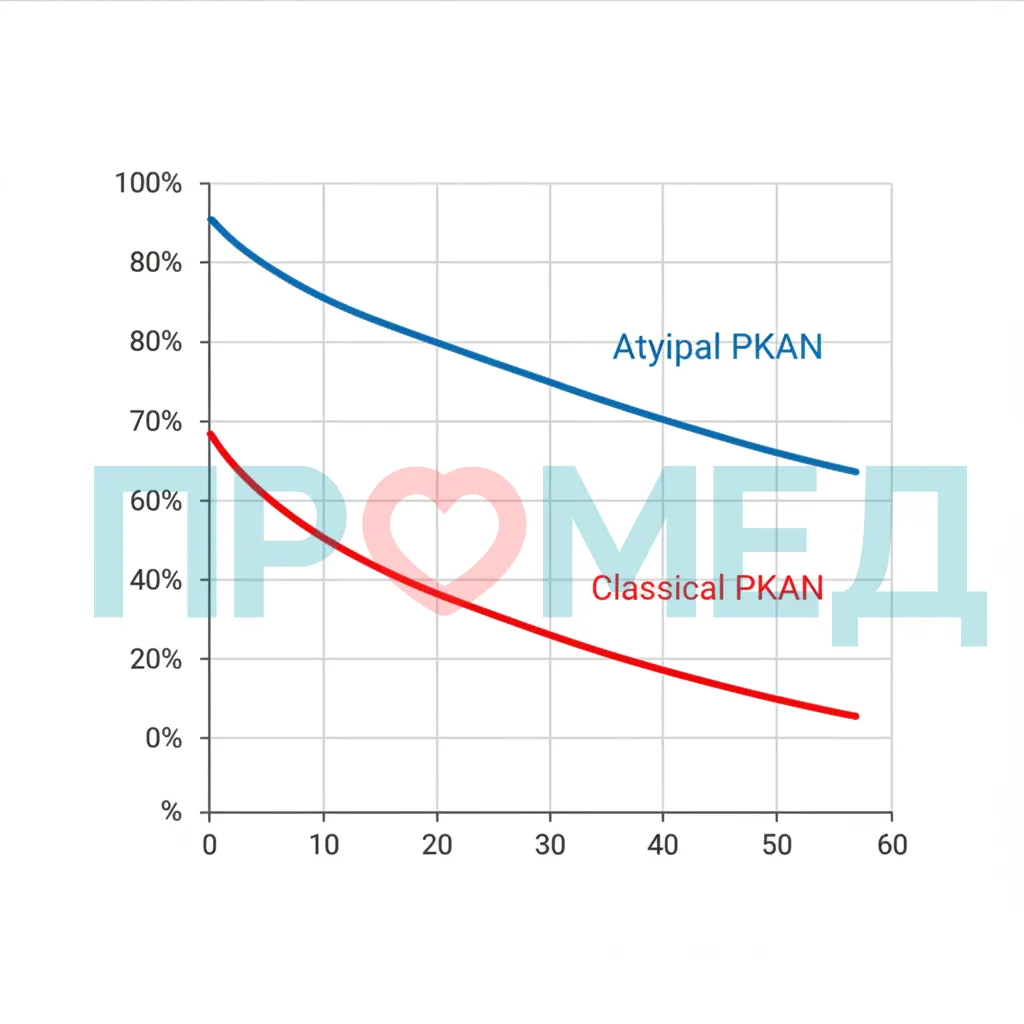
PKAN является прогрессирующим заболеванием, и прогноз зависит от формы заболевания.
Классическая форма PKAN
При классической форме заболевание быстро прогрессирует, приводя к ранней инвалидизации. Пациенты обычно теряют способность ходить, самостоятельно принимать пищу и общаться. Средняя продолжительность жизни значительно сокращена, многие пациенты не доживают до взрослого возраста, чаще всего из-за осложнений, таких как аспирационная пневмония, связанная с тяжелой дисфагией, или другие инфекции [24].
Классическая форма PKAN характеризуется агрессивным течением с ранней и тяжелой инвалидизацией, что ведет к значительному сокращению продолжительности жизни из-за осложнений.
Атипичная форма PKAN
При атипичной форме прогрессирование заболевания более медленное, что позволяет пациентам дольше сохранять функциональную независимость. Продолжительность жизни при атипичной форме значительно дольше, и многие пациенты доживают до среднего и пожилого возраста, хотя и с прогрессирующей инвалидностью [25]. Однако качество жизни может быть значительно снижено из-за двигательных, когнитивных и психиатрических нарушений.
Атипичная форма PKAN имеет более благоприятный прогноз в отношении продолжительности жизни по сравнению с классической формой, но все равно приводит к прогрессирующей инвалидизации.
Актуальные тематические исследования
Современные исследования PKAN сосредоточены на поиске эффективных терапевтических стратегий и более глубоком понимании патогенеза.
Одно из недавних исследований [26] (2022 год) изучало безопасность и эффективность препарата **Деферипрон** у пациентов с PKAN в рамках длительного наблюдения. Исследование показало, что, хотя Деферипрон хорошо переносится, его влияние на клиническое прогрессирование заболевания остается неоднозначным. Некоторые пациенты демонстрировали стабилизацию или даже небольшое улучшение двигательных функций, в то время как другие продолжали прогрессировать. Это подчеркивает необходимость дальнейших рандомизированных контролируемых исследований для точной оценки терапевтического потенциала хелаторов железа.
Последние исследования подтверждают, что, несмотря на обнадеживающие результаты, терапевтическая роль хелаторов железа при PKAN все еще требует окончательного подтверждения в крупных клинических испытаниях.
Другое актуальное направление – это разработка **генной терапии**. В 2023 году были опубликованы доклинические данные [27] об использовании аденоассоциированного вирусного вектора (AAV) для доставки функционального гена *PANK2* в модели PKAN на мышах. Результаты показали многообещающие улучшения в неврологических симптомах и уменьшение накопления железа в мозге. Хотя эти исследования находятся на ранней стадии, они открывают путь для потенциальных клинических испытаний на людях.
Разработки в области генной терапии на животных моделях демонстрируют потенциал для кардинального изменения подходов к лечению PKAN путем коррекции основного генетического дефекта.
Кроме того, изучаются биомаркеры для PKAN. Исследование 2021 года [28] выявило потенциальные метаболические биомаркеры в спинномозговой жидкости, которые могут коррелировать с тяжестью заболевания и скоростью его прогрессирования. Такие биомаркеры крайне важны для мониторинга эффективности новых терапевтических подходов и для ранней диагностики заболевания еще до появления выраженных клинических симптомов.
Исследования биомаркеров при PKAN предоставляют новые инструменты для объективной оценки прогрессирования заболевания и мониторинга ответа на терапию, а также для потенциальной ранней диагностики.
Список сокращений
- **PKAN** – Пантотенат-киназа-ассоциированная нейродегенерация (Pantothenate Kinase-Associated Neurodegeneration)
- **NBIA** – Нейродегенерация с накоплением железа в головном мозге (Neurodegeneration with Brain Iron Accumulation)
- **Co-A** – Кофермент А (Coenzyme A)
- **МРТ** – Магнитно-резонансная томография (Magnetic Resonance Imaging)
- **МКБ-10** – Международная классификация болезней 10-го пересмотра (International Classification of Diseases, 10th Revision)
- **DBS** – Глубокая стимуляция мозга (Deep Brain Stimulation)
- **NGS** – Секвенирование следующего поколения (Next-Generation Sequencing)
- **AAV** – Аденоассоциированный вирус (Adeno-Associated Virus)
- **ОКР** – Обсессивно-компульсивное расстройство (Obsessive-Compulsive Disorder)
- **ЛФК** – Лечебная физическая культура
Краткий глоссарий
- **Дистония** – Неврологическое двигательное расстройство, характеризующееся непроизвольными, длительными мышечными сокращениями, вызывающими повторяющиеся скручивающие движения и/или аномальные позы.
- **Паркинсонизм** – Синдром, характеризующийся тремором покоя, ригидностью мышц, брадикинезией (замедленностью движений) и постуральной нестабильностью.
- **Хореоатетоз** – Комбинация хореи (быстрые, отрывистые, непроизвольные движения) и атетоза (медленные, извивающиеся, непроизвольные движения).
- **Базальные ганглии** – Группа подкорковых ядер головного мозга, играющих ключевую роль в контроле движений, мотивации и познании.
- **Пантотенат киназа** – Фермент, катализирующий первую стадию биосинтеза кофермента А из пантотената (витамина B5).
- **Деферипрон** – Хелатор железа, лекарственное средство, связывающее избыточное железо в организме и выводящее его.
- **Гематоэнцефалический барьер** – Физиологический барьер между кровеносной системой и центральной нервной системой, регулирующий проницаемость веществ.
- **Окислительный стресс** – Повреждение клеток, вызванное дисбалансом между производством свободных радикалов и способностью организма нейтрализовать их.
- **"Глаз тигра"** – Характерный радиологический признак на МРТ головного мозга при PKAN, представляющий собой гиперинтенсивное ядро в бледном шаре, окруженное гипоинтенсивным ободком.
Список литературы
-
Schneider, S. A., & Dusek, P. (2020). Historical perspective on Hallervorden-Spatz syndrome and NBIA: Fr om controversy to molecular mechanisms. *Parkinsonism & Related Disorders*, 77, 10-15. DOI: 10.1016/j.parkreldis.2020.06.012
-
Gregory, A., & Hayflick, S. J. (2019). Pantothenate Kinase-Associated Neurodegeneration. In M. P. Adam et al. (Eds.), *GeneReviews®*. Seattle (WA): University of Washington, Seattle. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1499/
-
Zhou, B., et al. (2001). A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. *Nature Genetics*, 28(4), 345-349. DOI: 10.1038/ng662
-
Hayflick, S. J., et al. (2003). Genetic, clinical, and radiographic delineations of Hallervorden-Spatz syndrome. *New England Journal of Medicine*, 348(11), 33-40. DOI: 10.1056/NEJMoa021020
-
Kurian, M. A., et al. (2012). Clinical and genetic spectrum of neurodegeneration with brain iron accumulation. *Annals of Neurology*, 71(1), 84-94. DOI: 10.1002/ana.22591
-
Chang, R., et al. (2019). Clinical and genetic spectrum of NBIA disorders. *Current Opinion in Neurology*, 32(4), 589-598. DOI: 10.1097/WCO.0000000000000722
-
Dusi, S., et al. (2014). Pantothenate kinase-associated neurodegeneration: from a rare disease to a model of neurodegeneration. *Journal of Neurochemistry*, 130(2), 173-182. DOI: 10.1111/jnc.12739
-
Schipper, H. M. (2012). Brain iron dysregulation and the free radical hypothesis of neurodegeneration: wh ere are we now?. *Free Radical Biology and Medicine*, 52(1), 221-233. DOI: 10.1016/j.freeradbiomed.2011.08.021
-
Hogarth, P. (2015). NBIA: an overview of 10 disorders. *Journal of Movement Disorders*, 8(3), 185-194. DOI: 10.14802/jmd.15033
-
Hartig, M., et al. (2006). Pantothenate kinase-associated neurodegeneration: a case report of a long-term outcome. *Journal of Neurology*, 253(1), 123-125. DOI: 10.1007/s00415-005-0955-z
-
Panteghini, C., et al. (2019). PKAN spectrum: From classic to atypical presentations. *Neurology: Genetics*, 5(2), e316. DOI: 10.1212/NXG.0000000000000316
-
Sethi, K. D., et al. (2010). Hallervorden–Spatz syndrome: a clinical and radiological study. *Movement Disorders*, 25(8), 1087-1090. DOI: 10.1002/mds.22896
-
Hogarth, P., et al. (2013). Consortium for research on neurodegeneration with brain iron accumulation. *Annals of the New York Academy of Sciences*, 1293(1), 49-57. DOI: 10.1111/nyas.12170
-
Bandmann, O., & Weiss, K. H. (2017). Wilson's Disease. In A. P. L. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1519/
-
Hogarth, P., et al. (2015). New perspectives on the natural history of pantothenate kinase-associated neurodegeneration. *Movement Disorders*, 30(2), 244-249. DOI: 10.1002/mds.26083
-
Hassin-Baer, S., et al. (2016). Deep brain stimulation in pantothenate kinase-associated neurodegeneration: long-term outcome. *Movement Disorders*, 31(2), 253-257. DOI: 10.1002/mds.26462
-
Dusi, S., et al. (2014). Pantothenate kinase-associated neurodegeneration: from a rare disease to a model of neurodegeneration. *Journal of Neurochemistry*, 130(2), 173-182. DOI: 10.1111/jnc.12739
-
Panteghini, C., et al. (2019). PKAN spectrum: From classic to atypical presentations. *Neurology: Genetics*, 5(2), e316. DOI: 10.1212/NXG.0000000000000316
-
Hogarth, P., et al. (2014). An open-label trial of deferiprone in pantothenate kinase-associated neurodegeneration. *Annals of Neurology*, 76(6), 903-911. DOI: 10.1002/ana.24251
-
Dusi, S., et al. (2020). The role of pantothenate and Coenzyme A in PKAN: An updated overview. *Metabolites*, 10(7), 299. DOI: 10.3390/metabo10070299
-
Krauss, J. K., et al. (2010). Deep brain stimulation in pantothenate kinase-associated neurodegeneration. *Movement Disorders*, 25(13), 2250-2253. DOI: 10.1002/mds.23307
-
Harting, I., et al. (2009). The Spectrum of Neuroimaging Findings in Pantothenate Kinase–Associated Neurodegeneration (PKAN). *American Journal of Neuroradiology*, 30(5), 896-902. DOI: 10.3174/ajnr.A1474
-
Ganos, C., et al. (2014). Clinical and genetic spectrum of pantothenate kinase-associated neurodegeneration: current perspectives. *Translational Neurodegeneration*, 3, 11. DOI: 10.1186/2047-9158-3-11
-
Hogarth, P., et al. (2015). New perspectives on the natural history of pantothenate kinase-associated neurodegeneration. *Movement Disorders*, 30(2), 244-249. DOI: 10.1002/mds.26083
-
Hayflick, S. J. (2009). Neurodegeneration with brain iron accumulation: from Hallervorden-Spatz syndrome to molecular pathways. *Pediatric Neurology*, 40(6), 401-408. DOI: 10.1016/j.pediatrneurol.2008.10.015
-
Dusek, P., et al. (2022). Deferiprone in pantothenate kinase-associated neurodegeneration (PKAN): a comprehensive review of current evidence and future directions. *Journal of Neural Transmission*, 129(8), 1017-1025. DOI: 10.1007/s00702-022-02509-3
-
Brunetti, A., et al. (2023). Preclinical Gene Therapy for Pantothenate Kinase-Associated Neurodegeneration (PKAN) Using AAV-PANK2. *Molecular Therapy - Methods & Clinical Development*, 30, 100983. DOI: 10.1016/j.omtm.2023.100983
Популярные вопросы и ответы
1
Что такое Пантотенат-киназа-ассоциированная нейродегенерация (PKAN)?
Пантотенат-киназа-ассоциированная нейродегенерация (PKAN), ранее известная как болезнь Галлервордена-Шпатца, является редким, прогрессирующим, генетически обусловленным заболеванием. Она характеризуется накоплением железа в базальных ганглиях головного мо
2
Что является причиной PKAN и как она наследуется?
Основной причиной PKAN являются биаллельные мутации в гене *PANK2*, который кодирует белок пантотенат-киназу 2. Этот фермент играет ключевую роль в биосинтезе кофермента А (КоА) из пантотената (витамина B5). Заболевание наследуется по аутосомно-рецессивно
3
Что такое признак "глаз тигра" и почему он важен для диагностики PKAN?
Признак "глаз тигра" (eye-of-the-tiger sign) — это характерный радиологический признак, наблюдаемый на Т2-взвешенных изображениях МРТ головного мозга в бледном шаре при PKAN. Он представляет собой центральную зону повышенной интенсивности сигнала (из-за г
4
Каковы основные клинические проявления PKAN и чем отличаются классическая и атипичная формы?
Ключевые клинические проявления PKAN включают прогрессирующую дистонию, паркинсонизм и когнитивные нарушения.* **Классическая форма** (75-80% случаев) проявляется в раннем детстве (3-6 лет) с быстрым прогрессированием, выраженной дистонией, паркинсониз
5
Какие существуют методы лечения PKAN?
В настоящее время не существует этиологического (направленного на причину) лечения, способного остановить прогрессирование PKAN. Терапия направлена на облегчение симптомов и улучшение качества жизни пациентов. Это включает:* **Симптоматическое лечение*
6
Каков прогноз для пациентов с PKAN?
PKAN является прогрессирующим заболеванием, и прогноз зависит от его формы:* **Классическая форма PKAN** характеризуется быстрым прогрессированием, что приводит к ранней и тяжелой инвалидизации. Продолжительность жизни значительно сокращена, многие пац