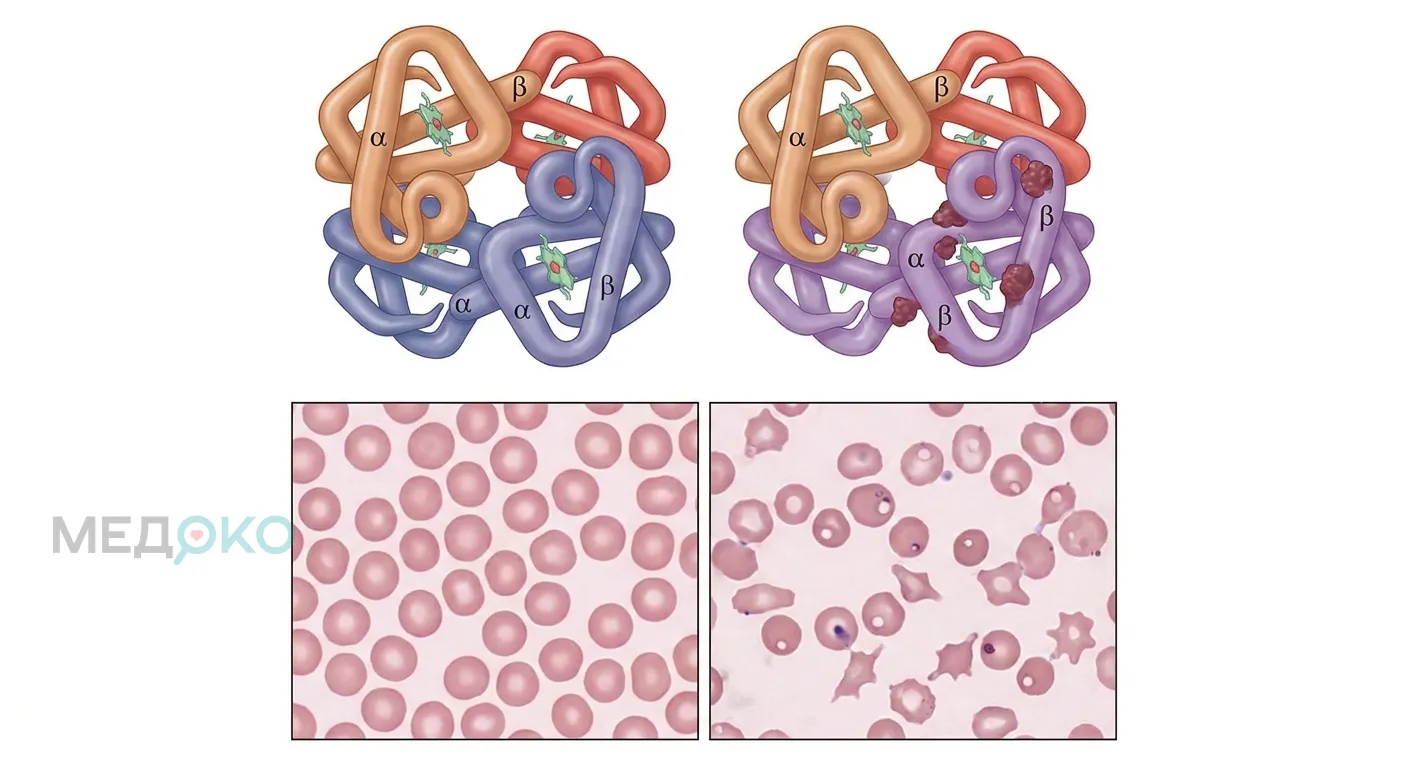
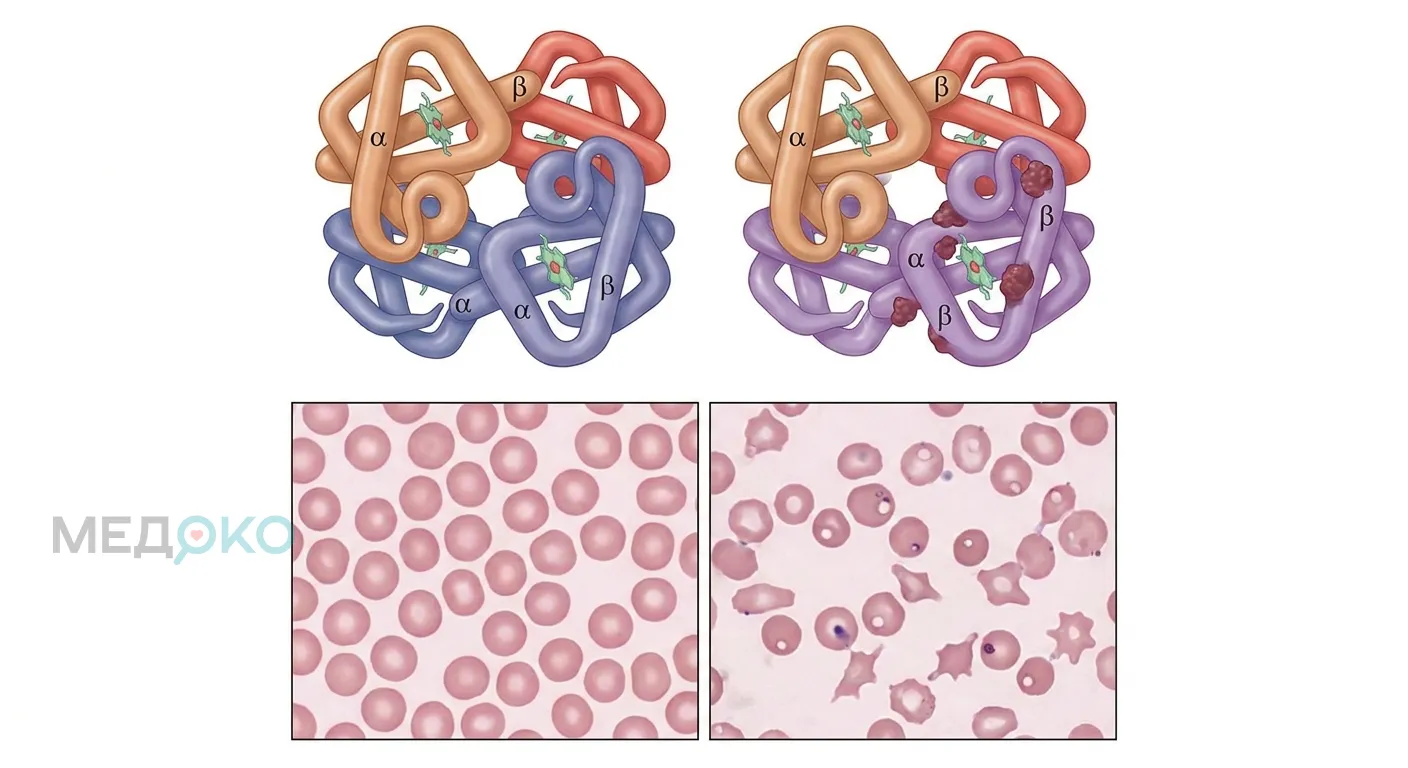
Альфа-талассемия у большинства носителей протекает легко, но при тяжёлых формах (болезнь гемоглобина H) могут возникать угрожающие состояния. Немедленно вызывайте скорую помощь или обращайтесь в приёмный покой, если у пациента наблюдаются: резкое побледнение кожи с выраженной слабостью (невозможность встать с кровати), внезапное пожелтение кожи и склер глаз, потемнение мочи до цвета крепкого чая (признаки острого гемолиза), одышка в состоянии покоя, учащенное сердцебиение на фоне головокружения или обморок. Беременным женщинам из группы риска требуется экстренное УЗИ при подозрении на задержку развития плода.
Альфа-талассемия за 30 секунд
Наследственное заболевание крови, при котором нарушается выработка альфа-цепей белка гемоглобина, что приводит к разрушению эритроцитов и развитию анемии.
Генетические мутации (делеции) в генах HBA1 и HBA2, передающиеся от родителей к детям.
D56.0 (Альфа-талассемия).
Заболевание врождённое и сохраняется на протяжении всей жизни.
Никогда не принимать препараты железа при выявлении анемии без предварительной сдачи анализа на ферритин (риск тяжелого отравления железом).
Гематолог, медицинский генетик.
Оглавление
Раздел 1 - Что такое альфа-талассемия
Альфа-талассемия - это генетическое заболевание крови, характеризующееся снижением или полным отсутствием синтеза альфа-глобиновых цепей [1]. Гемоглобин (белок, переносящий кислород) в норме состоит из двух альфа- и двух бета-цепей. При нехватке альфа-цепей баланс нарушается, эритроциты становятся мелкими (микроцитоз), бледными (гипохромия) и быстро разрушаются в селезенке, что приводит к анемии.
Место болезни среди схожих состояний уникально: это одно из самых распространенных моногенных заболеваний в мире, которое часто маскируется под банальный дефицит железа.
Сравнительная таблица: Альфа-талассемия vs похожие диагнозы
| Признак | Альфа-талассемия (малая форма) | Железодефицитная анемия (ЖДА) | Бета-талассемия (малая) |
|---|---|---|---|
| Глубина поражения | Генетический дефект альфа-цепей | Приобретенный дефицит железа | Генетический дефект бета-цепей |
| Уровень ферритина | Нормальный или повышенный | Снижен (менее 30 нг/мл) | Нормальный или повышенный |
| Число эритроцитов (RBC) | Повышено (более 5.0-5.5) | Снижено | Повышено |
| Электрофорез гемоглобина | Взрослый HbA в норме (при малых форм), у новорожденных есть Hb Barts | В норме | Повышен HbA2 (>3.5%) |
| Реакция на железо | Нет ответа (может навредить) | Быстрое улучшение показателей | Нет ответа |
Как отличить от железодефицитной анемии: Главный маркер - это соотношение уровня эритроцитов и объема эритроцита (MCV). При альфа-талассемии эритроцитов много, но они очень мелкие (индекс Ментцера часто меньше 13). При ЖДА эритроцитов мало, и они мелкие. Точку в диагнозе ставит анализ крови на ферритин и сывороточное железо [2].
Ключевые выводы:
- Альфа-талассемия - это врожденное нарушение синтеза гемоглобина, а не результат плохого питания или кровопотери.
- Главная опасность легких форм болезни - ошибочная диагностика дефицита железа и токсичное лечение препаратами железа.
- Отличить талассемию от ЖДА можно только с помощью специальных лабораторных тестов (ферритин, железо, электрофорез, генетика).
Причины и факторы риска
За синтез альфа-глобина отвечают четыре гена (по два от каждого родителя - HBA1 и HBA2), расположенные на 16-й хромосоме. Заболевание возникает из-за мутаций, чаще всего - делеций (выпадения участков ДНК), приводящих к «выключению» одного или нескольких из этих генов [3].
Механизм развития прост: чем меньше функционирующих генов, тем меньше альфа-цепей. Избыточные бета- или гамма-цепи образуют нестабильные соединения (гемоглобин H или гемоглобин Barts), которые повреждают мембрану эритроцита, вызывая его преждевременную гибель.
Факторы риска по группам:
| Группа факторов | Описание риска |
|---|---|
| Наследственные / Генетические | Наличие диагностированной талассемии или хронической неясной анемии у родителей, братьев или сестер. Риск передачи ребенку зависит от комбинации генов обоих родителей. |
| Этнические (Географические) | Происхождение из регионов эндемичных по малярии: Юго-Восточная Азия, Средиземноморье, Африка, Ближний Восток, Кавказ. Мутация закрепилась эволюционно, так как защищает носителей от тяжелого течения малярии [4]. |
| Поведенческие | Вступление в брак между близкими родственниками (повышает риск встречи двух патологических генов), планирование беременности без предварительного генетического скрининга в парах из группы риска. |
| Системные | Наличие других гемоглобинопатий (например, серповидноклеточной аномалии) в семье может приводить к сложным сочетанным формам болезни. |
Ключевые выводы:
- Болезнь не передается воздушно-капельным или контактным путем, заразиться ей невозможно - это исключительно генетический дефект.
- Тяжесть заболевания математически зависит от количества «сломанных» генов (от 1 до 4).
- Принадлежность к определенной этнической группе (например, жителям Азии или Кавказа) многократно повышает вероятность бессимптомного носительства.
Классификация и стадии
При альфа-талассемии не бывает «стадий» прогрессирования (как в онкологии). Тяжесть состояния определяется генетически с момента зачатия. Классификация базируется на количестве пораженных генов.
- Сроки: С рождения и на всю жизнь.
- Клиническая картина: Симптомы отсутствуют. Общий анализ крови в норме, иногда выявляется пограничное снижение объема эритроцитов (MCV).
- Тактика: Лечение не требуется. Важно только информирование пациента для планирования семьи.
- Сроки: С рождения.
- Клиническая картина: Легкая анемия, которая не влияет на качество жизни. Эритроциты мелкие (микроцитоз). Человек может чувствовать небольшую усталость при экстремальных нагрузках.
- Тактика: Избегать необоснованного приема железа. Наблюдение гематолога при беременности или тяжелых инфекциях.
- Сроки: Первые проявления - в раннем детстве.
- Клиническая картина: Анемия средней или тяжелой степени. Желтуха, увеличение селезенки (спленомегалия) и печени, деформации костей черепа (из-за разрастания костного мозга), склонность к образованию камней в желчном пузыре, задержка роста у детей [5].
- Тактика: Регулярное наблюдение. Прием фолиевой кислоты. При резком падении гемоглобина (гемолитический криз) - переливание крови. Иногда требуется удаление селезенки.
- Сроки: Развивается внутриутробно.
- Клиническая картина: Плод не может синтезировать функциональный гемоглобин. Развивается тяжелейшая гипоксия, сердечная недостаточность, массивные отеки (водянка).
- Тактика: Как правило, приводит к гибели плода внутриутробно или в первые часы после рождения. В редких случаях возможно спасение при помощи внутриутробных переливаний крови и последующей трансплантации костного мозга.
Ключевые выводы:
- Форма заболевания напрямую определяется количеством поврежденных генов: чем больше, тем тяжелее клиника.
- Пациенты с мутацией 1 или 2 генов ведут обычный образ жизни и нуждаются лишь в грамотном консультировании.
- Пациенты с мутацией 3 генов (болезнь HbH) требуют регулярного медицинского сопровождения и специфической терапии в периоды кризисов.
Симптомы и признаки
Клинические проявления напрямую зависят от формы заболевания (см. Раздел 3). Носители 1-2 мутаций обычно не предъявляют жалоб. Симптоматика разворачивается при болезни гемоглобина H (HbH).
Местные и специфические симптомы (при болезни HbH):
- Увеличение селезенки (спленомегалия): Пациент или врач может прощупать плотное образование в левом подреберье. Часто сопровождается чувством быстрого насыщения при еде (селезенка давит на желудок).
- Скелетные изменения: Поскольку костный мозг пытается компенсировать нехватку эритроцитов, он разрастается. Это приводит к утолщению костей черепа (выступающие скулы, башенный череп), искривлению зубов, хрупкости трубчатых костей.
- Желчнокаменная болезнь: Постоянное разрушение эритроцитов высвобождает много билирубина, который кристаллизуется в желчном пузыре [6].
Общие симптомы (следствие анемии и гипоксии):
- Постоянная, немотивированная слабость, снижение толерантности к физическим нагрузкам.
- Бледность кожи с землянистым или лимонно-желтым оттенком. Желтушность склер (глаз).
- Учащенное сердцебиение (тахикардия) и одышка при минимальных усилиях.
- У детей - отставание в физическом и половом развитии.
Обострение (гемолитический криз) может быть спровоцировано высокой температурой, тяжелой инфекцией или приемом некоторых лекарств (сульфаниламиды, противомалярийные). Признаки криза: резкое потемнение мочи, внезапная слабость вплоть до обморока, усиление желтухи, боли в левом боку. Требуется немедленная госпитализация.
Ключевые выводы:
- Симптоматика при легких формах стертая или отсутствует, диагноз ставится случайно по анализу крови.
- Хроническая усталость и бледность - самые частые, но не специфичные признаки, требующие дифференциальной диагностики.
- Потемнение мочи и желтуха на фоне инфекции - грозный признак активного распада эритроцитов.
Что делать: пошаговый план пациента
- Резкое падение артериального давления, обморок.
- Затрудненное дыхание, нехватка воздуха в покое.
- Острая боль в животе (особенно в левом подреберье) на фоне высокой температуры.
- Потемнение мочи (цвет колы или крепкого чая) за один-два дня.
Если вы получили анализ крови, где снижен гемоглобин и объем эритроцита (MCV), но чувствуете себя удовлетворительно, действуйте по плану:
- Не паникуйте и не покупайте железо. Это самая частая ошибка. Сначала нужно доказать дефицит железа.
- Сдайте дополнительный минимум: Ферритин, сывороточное железо, ОЖСС (общая железосвязывающая способность сыворотки), СРБ (С-реактивный белок - чтобы исключить ложное повышение ферритина из-за воспаления).
- Обратитесь к терапевту или гематологу. Если ферритин в норме или повышен, а анемия есть, врач заподозрит талассемию.
- Сдайте специфические анализы: Врач назначит электрофорез гемоглобина (капиллярный электрофорез) или генетический тест на мутации генов HBA1/HBA2.
- Пройдите УЗИ брюшной полости: Для оценки размеров селезенки, печени и проверки желчного пузыря на наличие камней.
- При подтверждении диагноза - обследуйте родственников. Особенно это важно для кровных братьев и сестер, а также при планировании беременности с партнером.
- Поддерживать сбалансированное питание, богатое витаминами группы В и фолиевой кислотой (зеленые листовые овощи, бобовые, орехи).
- Избегать переохлаждений и своевременно лечить ОРВИ (инфекции могут усиливать разрушение эритроцитов).
- Информировать любого нового врача (особенно хирурга или стоматолога) о наличии анемии.
- Принимать препараты железа или поливитамины с железом "для профилактики". При талассемии железо не усваивается эритроцитами должным образом, а накапливается во внутренних органах (печени, сердце), вызывая их токсическое повреждение (гемосидероз).
- Принимать сульфаниламидные антибиотики (например, Бисептол) без строгих показаний - они могут спровоцировать распад крови при болезни HbH.
Ключевые выводы:
- Первый шаг при микроцитарной анемии - сдача ферритина, а не прием железа.
- Только врач-гематолог может правильно расшифровать генетические и электрофоретические тесты.
- Самолечение железом при талассемии ведет к необратимому повреждению внутренних органов.
Диагностика
Постановка диагноза основывается на клиническом осмотре (оценка цвета кожи, пальпация живота), сборе семейного анамнеза и специфических лабораторных тестах.
Лабораторные анализы: что и зачем
- Клинический анализ крови (ОАК) с ретикулоцитами: Выявляет снижение гемоглобина, снижение MCV (средний объем эритроцита), снижение MCH (среднее содержание гемоглобина в эритроците). При этом количество эритроцитов (RBC) часто повышено >5.0х10^12/л. Уровень ретикулоцитов (молодых эритроцитов) обычно повышен, что говорит о попытке костного мозга компенсировать потери.
- Обмен железа (ферритин, сывороточное железо): Уровень ферритина при альфа-талассемии нормальный или повышенный [2].
- Электрофорез гемоглобина (или ВЭЖХ): Помогает разделить типы гемоглобина. Важно: у взрослых с малой альфа-талассемией электрофорез может быть абсолютно нормальным (в отличие от бета-талассемии). При болезни HbH выявляется специфический фракционный пик гемоглобина H [7].
- Молекулярно-генетическое тестирование (ДНК-анализ): Золотой стандарт. Метод ПЦР выявляет конкретные делеции генов HBA1 и HBA2. Точно определяет тип носительства (1, 2, 3 или 4 гена) [3].
Инструментальные методы:
- УЗИ органов брюшной полости (размеры селезенки, печени, камни в желчном).
- МРТ Т2* (Т2 со звездочкой) печени и сердца: назначается пациентам с болезнью HbH для оценки перегрузки органов железом (из-за переливаний крови или повышенного всасывания).
Дифференциальная диагностика: с чем путают
Чаще всего заболевание путают с железодефицитной анемией, анемией хронического заболевания или сидеробластной анемией.
Выбор специалиста и клиники:
Для генетического консультирования и ведения пациентов (особенно детей и беременных) выбирайте клиники, имеющие в штате профильных гематологов и возможность отправки крови на молекулярно-генетический анализ (ДНК).
Ключевые выводы:
- Общий анализ крови дает повод заподозрить талассемию (много мелких эритроцитов), но не подтверждает диагноз.
- Электрофорез гемоглобина может быть в норме у взрослых носителей альфа-талассемии, что часто сбивает врачей с толку.
- ДНК-анализ - единственный абсолютно точный метод подтверждения диагноза и определения формы болезни.
Методы лечения
Лечение напрямую зависит от генетической формы болезни. Полностью вылечить талассемию медикаментами невозможно, терапия направлена на поддержание качества жизни и предотвращение осложнений.
Таблица: Тактика ведения по формам заболевания
| Форма (Кол-во мутаций) | Консервативное лечение | Хирургическое лечение | Системное/Специфическое |
|---|---|---|---|
| Носительство (1-2 гена) | Не требуется. Обычный образ жизни. | Не показано. | Не показано. |
| Болезнь HbH (3 гена) | Фолиевая кислота (поддержка кроветворения). Хелаторная терапия (препараты, выводящие лишнее железо). Трансфузии (переливания) эритромассы - только при кризах [8]. | Спленомегалия: удаление селезенки (спленэктомия) при гигантских размерах или выраженном разрушении крови. Холецистэктомия (при камнях). | Трансплантация гемопоэтических стволовых клеток (костного мозга) в редких тяжелых случаях. |
| Болезнь Hb Barts (4 гена) | Экстренные внутриутробные переливания крови (спасение жизни плода). | - | Трансплантация стволовых клеток от совместимого донора (единственный шанс на излечение). |
Показания к госпитализации:
Госпитализация в гематологическое отделение требуется при болезни гемоглобина H в случае гемолитического криза (выраженная желтуха, падение гемоглобина ниже 70 г/л), развития тяжелой инфекции или необходимости проведения хирургической операции (удаление селезенки).
Критерии успешного лечения (для тяжелых форм):
Удержание уровня гемоглобина на показателях, обеспечивающих нормальный рост и развитие ребенка (обычно 90-100 г/л). Контроль уровня ферритина в безопасной зоне с помощью хелаторной терапии (предотвращение перегрузки железом).
Ключевые выводы:
- Большинство пациентов с альфа-талассемией (1-2 мутации) не нуждаются в лечении вообще.
- Пациентам с болезнью HbH (3 мутации) могут потребоваться периодические переливания крови и препараты для выведения токсичного железа.
- Единственный метод радикального излечения тяжелых форм - пересадка костного мозга, сопряженная с высокими рисками.
Особые группы пациентов
Дети
У детей с малым носительством симптомов нет. Дети с болезнью HbH требуют пристального внимания педиатра и гематолога. Хроническая гипоксия без лечения может привести к задержке роста, полового созревания и деформациям лицевого скелета. Срочный визит к врачу нужен при любых признаках необъяснимой вялости, желтухи на фоне вирусной инфекции.
Беременные
Это критическая группа. Риски делятся на две категории:
- Для матери-носительницы: Во время беременности физиологическая потребность в эритроцитах растет, анемия может усугубиться. Обычные витамины для беременных содержат железо - их прием нужно строго согласовывать с врачом, опираясь на уровень ферритина!
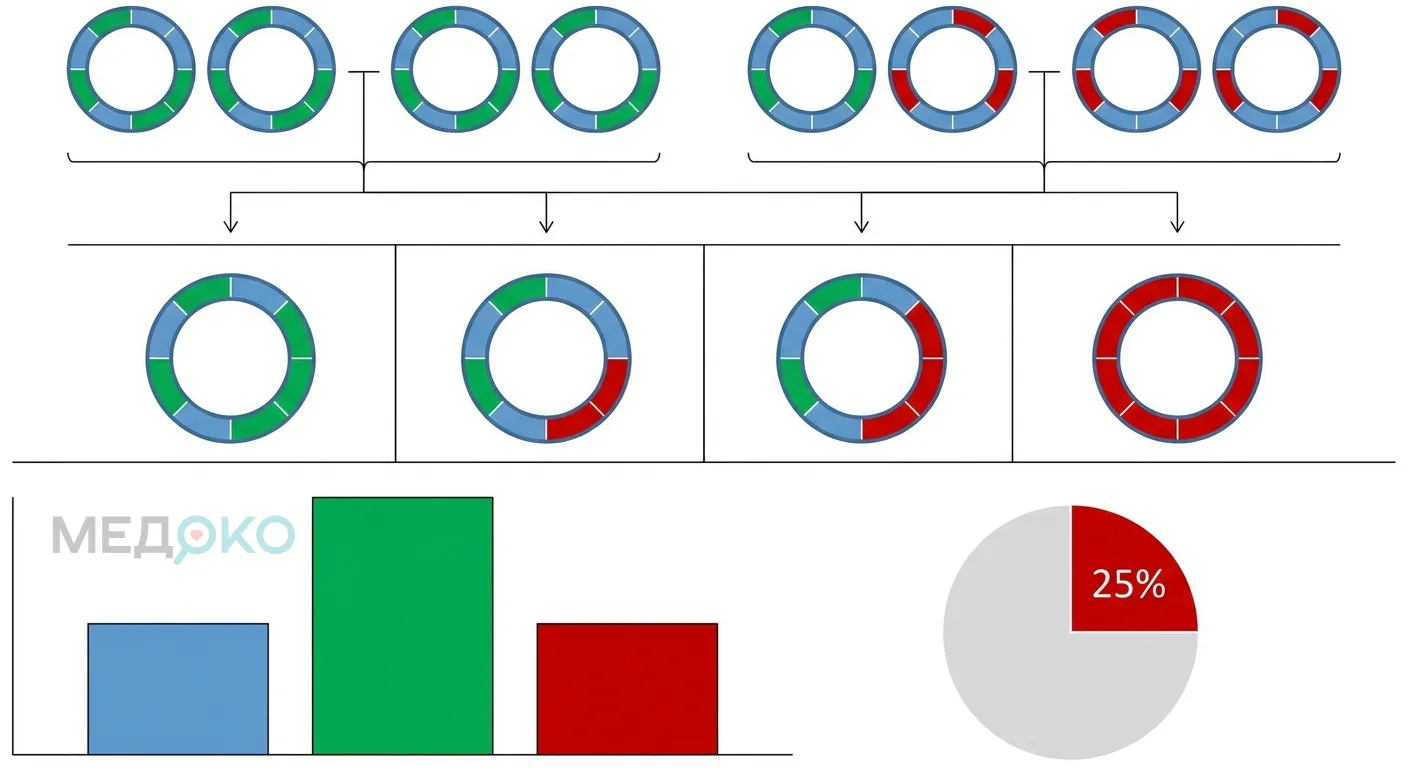
- Для плода: Если оба родителя имеют мутации альфа-глобина, есть 25% риск зачатия ребенка с тяжелой формой (синдром водянки плода Hb Barts). Это смертельное состояние для плода, которое также вызывает тяжелые осложнения у беременной (тяжелая преэклампсия, гипертония, кровотечения) [9]. Наблюдение в перинатальном центре обязательно.
Пожилые
У пожилых пациентов с малыми формами талассемии могут возникать трудности при диагностике других возрастных анемий (например, на фоне скрытых кровотечений из кишечника). Талассемия "маскирует" развитие ЖДА. Врачу приходится ориентироваться только на снижение ферритина.
Ключевые выводы:
- Беременность в парах с талассемией требует генетического скрининга до зачатия или инвазивной пренатальной диагностики на ранних сроках.
- Водянка плода опасна не только для ребенка, но и угрожает жизни матери.
- Детям с болезнью HbH необходим строгий контроль для предотвращения задержки развития и костных деформаций.
Частые ошибки пациентов
Легкие формы талассемии протекают доброкачественно, но неправильные действия пациентов могут нанести серьезный вред здоровью.
Механизм вреда: При талассемии железо из пищи всасывается даже активнее, чем в норме. Организм не может использовать его для строительства полноценного гемоглобина из-за генетического дефекта. Лишнее железо откладывается в печени и сердце (гемосидероз), приводя к циррозу и сердечной недостаточности.
Механизм вреда: Если один партнер знает о своей "малой талассемии", а второй не обследован, пара может столкнуться с рождением ребенка с несовместимой с жизнью водянкой плода (Hb Barts) или тяжелой болезнью HbH.
Механизм вреда: Из-за ускоренного распада и синтеза новых эритроцитов костный мозг стремительно расходует фолиевую кислоту. Ее дефицит может вызвать мегалобластный криз (глубокое падение гемоглобина).
Механизм вреда: Некоторые препараты (аспирин в высоких дозах, сульфаниламиды) вызывают окислительный стресс эритроцитов, провоцируя их массированное разрушение у пациентов с болезнью HbH.
Механизм вреда: Это не поможет исправить сломанный ген, но обеспечит ненужную избыточную нагрузку железом на желудочно-кишечный тракт.
Ключевые выводы:
- Низкий гемоглобин не всегда равно "дефицит железа".
- Планирование беременности у носителей мутации требует участия двух партнеров.
- Диета при талассемии не способна повысить уровень гемоглобина до нормы.

Профилактика
Первичная профилактика (предотвращение болезни):
Поскольку заболевание генетическое, предотвратить само появление мутации невозможно. Единственный путь - профилактика рождения детей с тяжелыми формами болезни в семьях риска. Это достигается путем:
- Медико-генетического консультирования пар до зачатия.
- Скрининга (ОАК + электрофорез гемоглобина + ДНК-диагностика) населения в эндемичных регионах (Кавказ, Азия) [4].
- Преимплантационной генетической диагностики (при ЭКО отбираются эмбрионы без тяжелых мутаций).
- Пренатальной диагностики (биопсия ворсин хориона или амниоцентез на 11-16 неделе беременности) [9].
Вторичная профилактика (предотвращение осложнений):
Для пациентов с болезнью HbH:
- Ежегодное УЗИ органов брюшной полости.
- Контроль уровня ферритина крови не реже 1 раза в год.
- Регулярный прием фолиевой кислоты по назначению врача.
- Вакцинация (от пневмококка, менингококка, гемофильной палочки, ежегодная от гриппа) - особенно важна для пациентов, которым удалили селезенку.
Показания к диспансерному наблюдению:
Пациенты с бессимптомным носительством (1-2 гена) с учета гематолога снимаются. Пациенты с болезнью HbH наблюдаются пожизненно: осмотр гематолога 2-4 раза в год в зависимости от тяжести течения.
Ключевые выводы:
- Основной метод борьбы с тяжелой талассемией - генетическое обследование пар перед беременностью.
- При подтвержденной болезни HbH крайне важна защита от инфекций, включая своевременную вакцинацию.
- Регулярный мониторинг уровня ферритина защищает органы от перегрузки железом.
FAQ: Частые вопросы
Нет, консервативного излечения не существует, так как это генетическая особенность. Тяжелые формы (HbH) в редких случаях можно вылечить трансплантацией костного мозга, но эта процедура имеет высокие риски и проводится по строгим показаниям [8]. Малые формы лечения не требуют.
Да. Если ваш партнер здоров (у него нет мутаций альфа-глобина), ваши детей будут либо полностью здоровы, либо станут такими же бессимптомными носителями, как и вы [3]. Риск тяжелой болезни возникает только если мутации есть у обоих родителей.
Для носителей легкой формы строгих ограничений нет. При болезни HbH (и других формах с перегрузкой железом) рекомендуется ограничить потребление продуктов, богатых легкоусвояемым (гемовым) железом - красного мяса, субпродуктов. Не рекомендуется пить алкоголь (особенно красное вино), так как он усиливает всасывание железа. Полезно пить черный чай вместе с едой (танины снижают усвоение железа).
При бессимптомном носительстве (1-2 мутации) можно заниматься любым спортом без ограничений. При болезни гемоглобина H (3 мутации) тяжелые физические нагрузки могут вызывать одышку и усталость; спорт разрешен в щадящем режиме, без экстремального перенапряжения. Контактные виды спорта могут быть запрещены при увеличенной селезенке (риск разрыва).
Потому что в ОАК при талассемии снижен объем эритроцита (MCV) и гемоглобин - точно такая же картина наблюдается при самом распространенном в мире дефиците железа (ЖДА). Если врач не назначает анализ на ферритин для проверки запасов железа, происходит диагностическая ошибка [2].
Нет. Генетический набор человека не меняется в течение жизни. Если у вас выявлена делеция двух генов (альфа-талассемия трейт), она никогда не "превратится" в болезнь гемоглобина H (три гена).
В большинстве стран, включая РФ, наличие гемолитический анемии (включая талассемию) является противопоказанием к донорству крови, так как ваши эритроциты имеют дефект мембраны и меньший срок жизни. Однако вы можете уточнить возможность донорства плазмы на местной станции переливания.
Источники и литература
- World Health Organization. Thalassaemia. (дата обращения: 18.02.2026).
- Клинические рекомендации Минздрава РФ. Железодефицитная анемия. (дата обращения: 18.02.2026).
- Centers for Disease Control and Prevention (CDC). Thalassemia. (дата обращения: 18.02.2026).
- Origa R. Alpha-Thalassemia. GeneReviews. (дата обращения: 18.02.2026).
- Fucharoen S, Viprakasit V. Hemoglobin H disease: clinical course and disease modifiers. Blood. (дата обращения: 18.02.2026).
- American Society of Hematology. Thalassemia. (дата обращения: 18.02.2026).
- Viprakasit V, Ekwattanakit S. Clinical classification, screening and diagnosis for thalassemia. Orphanet J Rare Dis. (дата обращения: 18.02.2026).
- Thalassemia International Federation (TIF). Guidelines for the Management of Transfusion Dependent Thalassaemia. (дата обращения: 18.02.2026).
- ACOG Practice Bulletin. Hemoglobinopathies in Pregnancy. (дата обращения: 18.02.2026).
Автор: медицинский редактор, портал Med-Oko.
Материал подготовлен в соответствии с редакционной политикой сайта.
Материал пересматривается при обновлении источников, изменении клинических подходов или не реже одного раза в год.
Дата последнего пересмотра: 18.02.2026 г.
Материал предназначен для информирования и не является диагнозом, назначением лечения или заменой очного осмотра. При выраженной бледности, внезапной слабости, одышке в покое или появлении желтухи на фоне сниженного гемоглобина обратитесь к врачу.