Синдром Аперта: главное о заболевании
Определение болезни
Синдром Аперта (Акроцефалосиндактилия типа I) - это редкое врожденное генетическое заболевание, которое характеризуется тремя основными признаками:
- Краниосиностоз: Преждевременное сращение швов черепа, приводящее к его деформации (часто "башенной" формы).
- Гипоплазия средней трети лица: Недоразвитие лицевых костей, из-за чего глаза кажутся выпученными и широко расставленными.
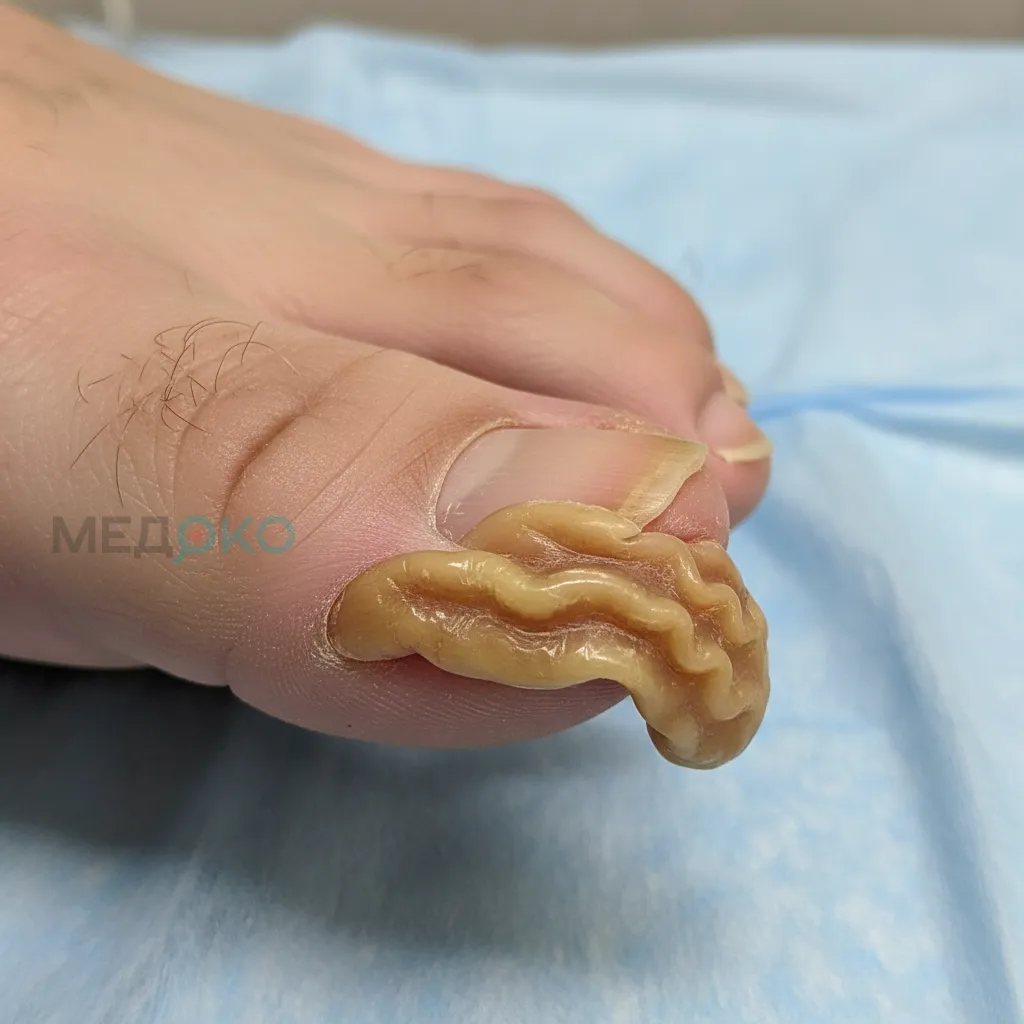
- Синдактилия: Сложное симметричное сращение пальцев на руках и ногах, создающее вид "руки-варежки".
Этиология и патогенез
Причиной заболевания являются мутации в гене FGFR2, который отвечает за сигналы, контролирующие рост и развитие костной ткани. В результате мутации рецептор становится постоянно активным, что приводит к ускоренному и преждевременному окостенению черепных швов и нарушению разделения пальцев во время внутриутробного развития.
Более 98% случаев синдрома Аперта вызваны двумя специфическими мутациями в гене FGFR2: Ser252Trp (S252W) и Pro253Arg (P253R). Большинство случаев являются результатом спонтанных мутаций (de novo), риск которых повышается с увеличением возраста отца.
Клиническая картина
Клинические проявления затрагивают множество систем организма:
- Череп и лицо: Акроцефалия ("башенный череп"), высокий и широкий лоб, плоский затылок, широко расставленные и выпученные глаза (экзофтальм), недоразвитая верхняя челюсть.
- Конечности: Симметричное сращение 2, 3 и 4 пальцев кистей и стоп, часто с вовлечением большого пальца и мизинца. Это серьезно ограничивает хватательную функцию.
- Другие системы: Возможны умственная отсталость разной степени, проблемы со зрением (косоглазие) и слухом (тугоухость), обструктивное апноэ сна, врожденные пороки сердца.
Диагностика
Диагностика синдрома Аперта включает несколько этапов:
- Пренатальная диагностика: Заподозрить синдром можно на УЗИ во втором-третьем триместре по аномальной форме черепа и синдактилии.
- Постнатальная диагностика: Диагноз ставится на основе характерных внешних признаков после рождения.
- Подтверждение: "Золотым стандартом" является молекулярно-генетический анализ для выявления мутаций в гене FGFR2. Для планирования операций используется КТ черепа с 3D-реконструкцией.
Ключевым моментом является дифференциальная диагностика с другими краниосиностозными синдромами.
Сравнительная таблица синдромов
| Признак |
Синдром Аперта |
Синдром Крузона |
Синдром Пфайффера |
| Аномалии кистей и стоп |
Тяжелая синдактилия ("рука-варежка") |
Нормальное строение |
Широкие и отклоненные большие пальцы |
| Интеллект |
Часто снижен |
Обычно нормальный |
Часто снижен |
| Основной ген |
FGFR2 |
FGFR2 |
FGFR1, FGFR2 |
Лечение
Лечение синдрома Аперта - это многоэтапный и пожизненный процесс, требующий участия команды врачей (нейрохирурги, ортопеды, офтальмологи, логопеды и др.).
- Хирургия черепа: Проводится в несколько этапов, начиная с 3-6 месяцев жизни. Цель - увеличить объем черепа для нормального роста мозга, снизить внутричерепное давление и скорректировать деформации лица.
- Хирургия конечностей: Многоэтапные операции по разделению пальцев для формирования функциональной кисти, позволяющей выполнять захват.
- Лечение сопутствующих патологий: Установка шунтов при гидроцефалии, коррекция зрения и слуха, ортодонтическое лечение.
Когда нужно обратиться к врачу
Необходимо немедленно обратиться к педиатру и генетику, если у новорожденного ребенка наблюдаются следующие признаки:
- Явно неправильная, вытянутая вверх или конусообразная форма головы.
- Полное или частичное сращение пальцев на руках или ногах.
- Сильно выпученные и/или широко расставленные глаза.
Ранняя диагностика критически важна для своевременного начала хирургического лечения и предотвращения тяжелых осложнений, таких как повреждение мозга из-за высокого внутричерепного давления.
Читать полную статью
Поделиться кратким содержанием
Популярные вопросы и ответы
1
Это наша вина, что у ребенка синдром Аперта?
Нет, это не ваша вина. В подавляющем большинстве случаев синдром Аперта возникает из-за новой, случайной мутации в гене ребенка, которая не была унаследована от родителей. Это спонтанное генетическое событие.
2
Можно ли полностью вылечить синдром Аперта?
Поскольку синдром Аперта является генетическим заболеванием, устранить его первопричину (мутацию в гене) на данный момент невозможно. Однако современная медицина предлагает множество хирургических и реабилитационных методов, направленных на коррекцию проя
3
Все ли дети с синдромом Аперта имеют умственную отсталость?
Нет, не все. Интеллектуальное развитие при синдроме Аперта может варьироваться в широких пределах – от нормы до различных степеней задержки. Своевременное хирургическое лечение, направленное на снижение внутричерепного давления, и комплексная реабилитация
4
Почему нельзя сделать все необходимые операции сразу в раннем детстве?
Хирургические вмешательства планируются поэтапно в соответствии с ростом и развитием ребенка. Операции на черепе в младенчестве необходимы для создания пространства растущему мозгу. Коррекция лица проводится позже, когда кости лицевого скелета достигают о
5
Сможет ли мой ребенок в будущем иметь здоровых детей?
Синдром Аперта передается по аутосомно-доминантному типу. Это означает, что у человека с этим синдромом риск передачи мутантного гена каждому своему ребенку составляет 50%. Современные репродуктивные технологии могут предложить варианты планирования семьи
6
Если у нас уже есть ребенок с синдромом Аперта, какой риск, что следующий ребенок родится с таким же диагнозом?
Если у родителей нет мутации (что подтверждается генетическим анализом), то риск рождения второго ребенка с этим же синдромом крайне низкий и практически не отличается от общепопуляционного. Для точной оценки рекомендуется консультация генетика.